Metabolically Active Adrenal Masses
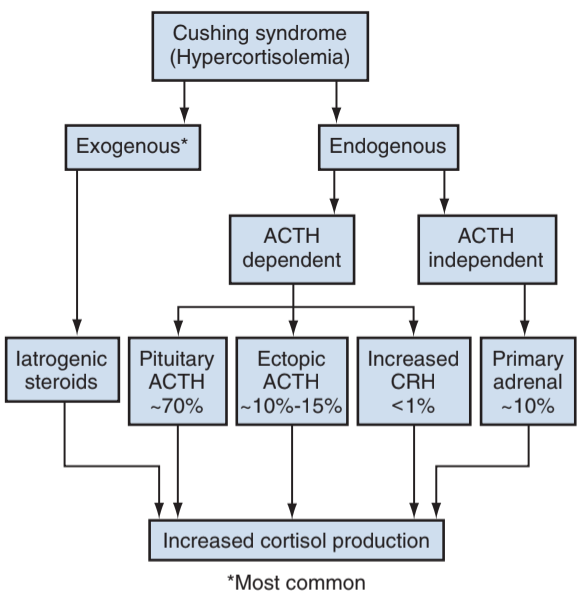
Causes of Cushing syndrome, from Campbell's
Hypercortisolism (Cushing Syndrome)
Causes
- Exogenous: most common cause due to treatment of other conditions
- ACTH dependent: 80% due to pituitary tumor (Cushing disease), ectopic ACTH production usually secondary to malignancy
- ACTH independent: due to adrenal adenomas, carcinomas, or hyperplasia
Urologic Indications for Hypercortisol Workup
- Hypogonadal hypogonadism: consider if low T/gonadotropins and ED or libido issues
- Stones: seen in up to 50% Cushing syndrome patients, increased stone risk even after treatment for Cushing
Workup
- Low dose dexamethasone suppression test: nighttime administration of dexamethasone should suppress ACTH and therefore cause decreased cortisol levels the next morning, but patients with Cushing syndrome will not have suppressed cortisol levels due to lack of negative feedback
- Identify source: check ACTH level, low level indicates primary adrenal, high level indicates pituitary or ectopic source
- Low ACTH: check abdominal imaging, consider exogenous sources if no adrenal lesions noted
- High ACTH and no obvious source screening imaging is non-diagnostic - measure ACTH levels in the inferior petrosal sinus vs peripheral plasma after CRH stimulation - high ratio indicates pituitary source whereas normal ratio indicates ectopic source
Autonomous Cortisol Secretion
- Definition: indeterminate range 1.8-5ug/dL
- Low (< 1%) risk for progression to overt Cushing syndrome
- Screening: assess for HTN, HLD, DM, vertebral fractures - all can be exacerbated/worsened by cortisol excess
- Surgery: consider only if clearly from adrenal adenoma and concern that low elevation of cortisol is causing undesired side effects
Treatment
- Exogenous steroids: taper source to prevent withdrawal
- Pituitary source: transsphenoidal surgical resection, with 60-80% cure and 25% relapse
- Solitary adrenal lesion: unilateral adrenalectomy
- Bilateral adrenal lesions or resistant to pituitary treatment: bilateral adrenalectomy
- Ectopic source: resection of ectopic source or bilateral adrenalectomy if unable to identify source (up to 35% patients)
- Medical therapies: metyrapone, aminoglutethimide, ketoconazole, etomidate, mitotane
- Nelson syndrome: pituitary adenoma growth after bilateral adrenalectomy, seen in 8-29% patients
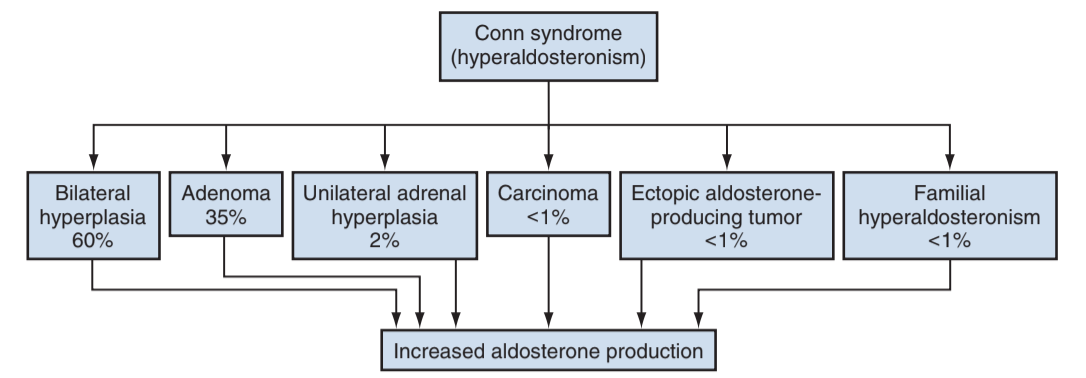
Causes of Conn syndrome, from Campbell's
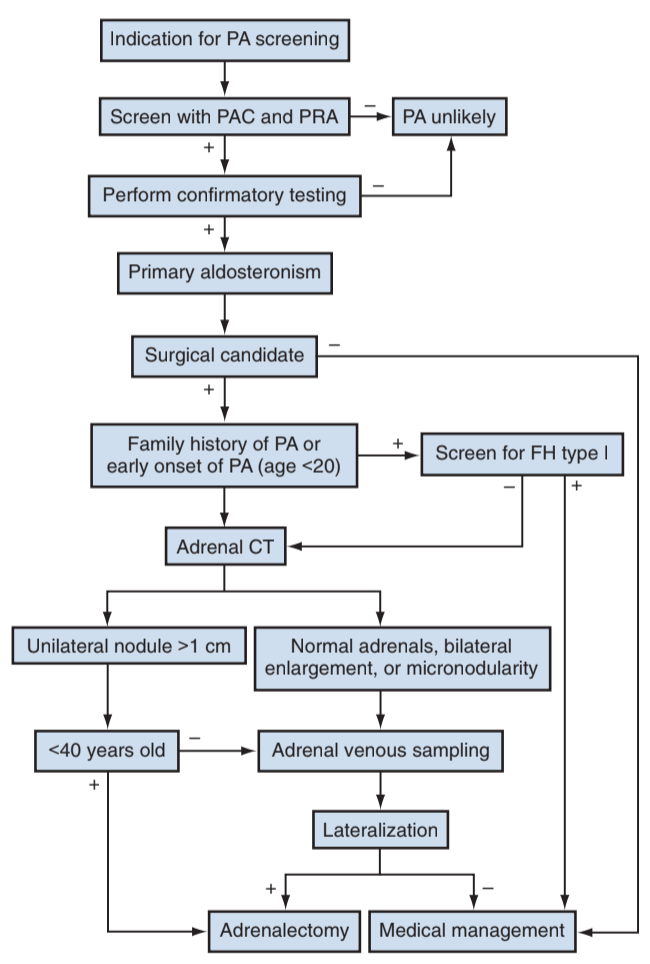
Primary hyperaldosteronism workup, from Campbell's
Hyperaldosteronism (Conn Syndrome)
Causes
- Idiopathic hyperplasia: no obvious adenoma, less likely to cause hypokalemia
- Adenoma: more likely to cause hypertension and hypokalemia
- Rare causes: adrenal carcinomas, ectopic production, familial causes
Workup
- Screening: usually found during workup for refractory hypertension, prevalence 2-13% depending on severity of blood pressure
- Hypokalemia: seen in 17-50% patients, also may report myalgias, polyuria, headaches
- Stop spironolactone and other receptor blockers 4 weeks prior to testing
- Hormonal screen: check morning (8-10AM) aldosterone and renin concentrations, calculate ratio (aldosterone/renin), perform confirmatory test if positive due to testing variability
- No need for subtype differentiation if patient is not a surgical candidate
- Genetic screening: indicated if family hx aldosteronism, age of onset < 20yo, or family hx CVAs
- CT imaging: adenomas < 10 HU, 20% less than 1cm
- Adrenal vein sampling: lateralizes source of aldosterone if not obvious on imaging, expected lateralizing ratio is > 2:1-4:1, not needed if < 40 and clear unilateral adenoma, or patients with ACC
Treatment
- Adrenalectomy: for lateralizing aldosterone secretion
- Ensure Mg + Phos repletion, normalize fluid status, BP control, and stress dose cortisol
- Medical management: spironolactone (25->400mg/d) and eplerenone (25->100mg/d) for patients who are not surgical candidates
Adrenal Insufficiency (Addison Disease)
Causes
- Autoimmune: most common cause in developed countries, may be seen in autoimmune polyendocrine syndrome
- Tuberculosis: most common cause in developing countries
- Sheehan syndrome: peripartum pituitary failure
- Others: hemorrhage, amyloidosis, sarcoidosis, hemochromatosis
Evaluation and Management
- Diagnosis: often made clinically, but confirmed with morning cortisol and ACTH, plus aldosterone and renin
- Adrenal crisis: hypotension unresponsive to fluid resuscitation, abdominal pain, nausea, vomiting, fever
- Mineralocorticoid deficiency: only seen in primary Addison disease
- Hyperpigmentation: ACTH stimulation also increases peptides that stimulate skin melanin production
- Treatment: glucocorticoids (+/- mineralocorticoids), give stress dose steroids prior to surgery
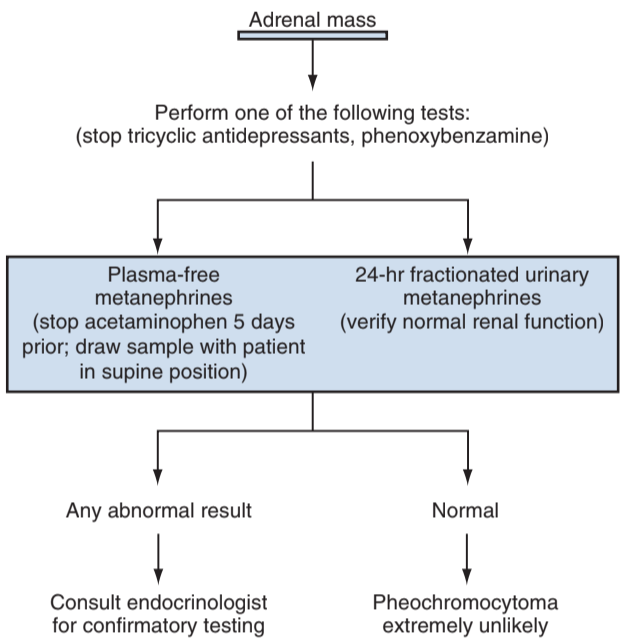
PCC diagnosis, from Campbell's
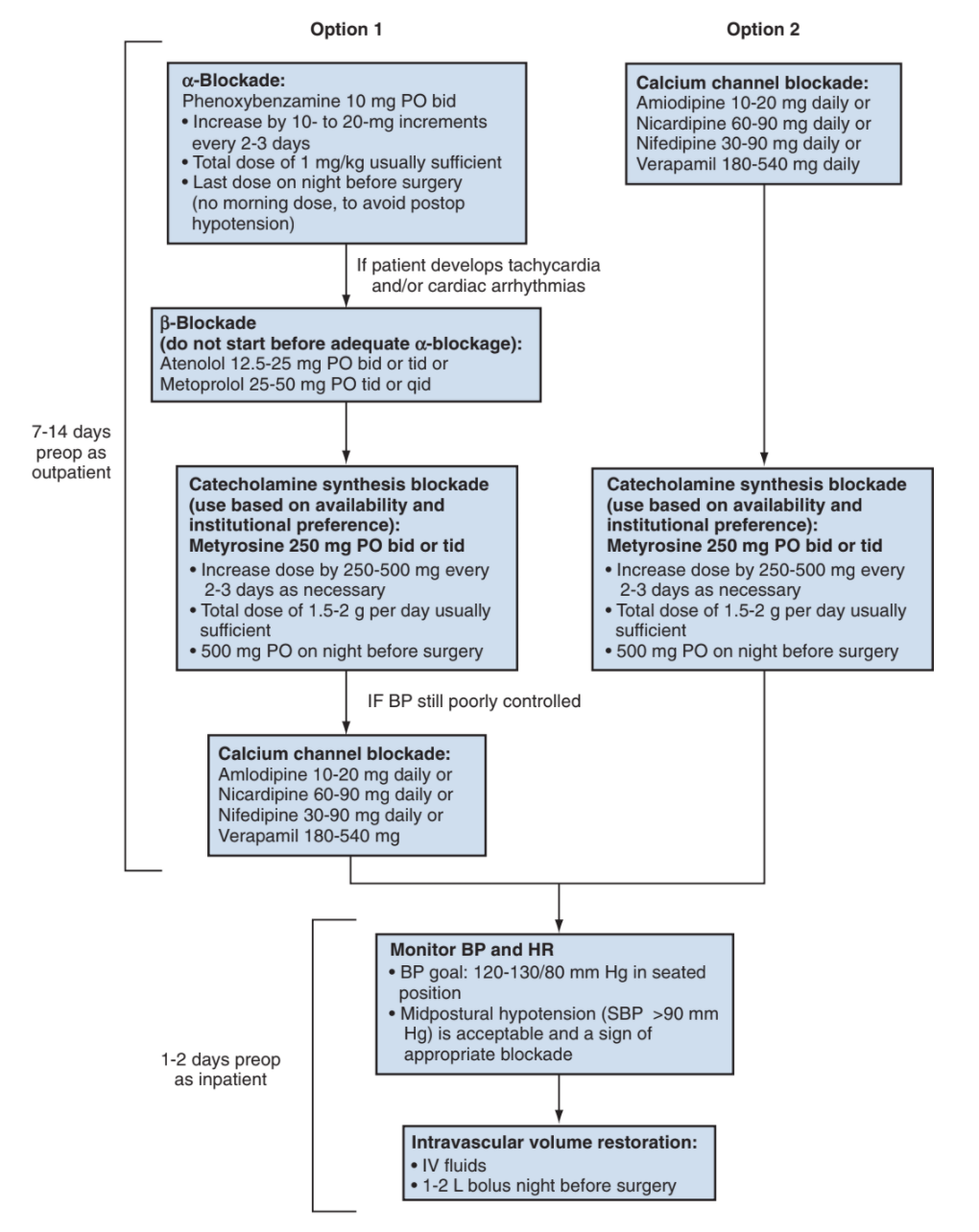
Perioperative management of PCC, from Campbell's
Pheochromocytoma (PCC) and Paragangliomas (Extra-adrenal PCCs)
Symptoms/Presentation
- Classic triad: headache, episodic diaphoresis, tachycardia
- Underlying cause in 0.5% HTN cases
- Epinephrine-producing tumors: may cause syncopal or hypotensive episodes due to B2 vasodilation
- Norepinephrine-producing tumors: cause hypertension and sweating due to A1 vasoconstriction
- Malignant PCC defined by presence of clinical metastasis
- Familial syndromes: seen in 1/3 cases, including MEN2A, MEN2B, VHL, NF1, PGL-1, PGL-4
Workup
- CT imaging: 10-35HU without contrast, do not exhibit rapid contrast washout
- PET: 18F-FDG is gold standard for staging, some centers use 68Ga-DOTATATE
- Metaiodobenzylguanidine (MIBG): analog of norepinephrine, used for staging, being replaced with PET
- Metanephrines: are metabolites of catecholamines, produced at a steady rate, and are more accurately detected than catecholamine episodic surges
- Genetic screening: obtain if family hx, < 50yo, multiple tumors, malignancy, or bilateral disease
Treatment/Follow-Up
- Laparoscopic adrenalectomy: standard of care, although open approach may be required
- a-blockade: start phenoxybenzamine 7-14 days preop, dose 10mg BID, titrate up to maintain a systolic BP of 120-130
- B-blockade: can be used to prevent tachycardia and arrhythmias, but should never be started before a-blockade (can exacerbate HTN)
- Malignant PCC: surgery is palliative, chemotherapy consists of cyclophosphamide + vincristine + dacarbazine (CVD)
- Recurrence: up to 16% at 10yrs, surveill with annual lab screening, imaging only based on lab results
References
- AUA Core Curriculum
- Fassnacht, Martin, et al. "Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the European network for the study of adrenal tumors." European journal of endocrinology 175.2 (2016): G1-G34.
- Lim, SK. and K. Ho Rha. "Surgery of the Adrenal Glands." Campbell-Walsh Urology 12 (2020).
- Maas, Marissa, et al. "Discrepancies in the recommended management of adrenal incidentalomas by various guidelines." The Journal of Urology 205.1 (2021): 52-59.
- Kutikov, A., P. L. Crispen, and R. G. Uzzo. "Pathophysiology, evaluation, and medical management of adrenal disorders." Campbell-Walsh Urology 12 (2020).